Announcing lime - Explaining the predictions of black-box models
![]()
I’m very pleased to announce that lime has been released on CRAN. lime is an implementation of the model prediction explanation framework described in “Why Should I Trust You?”: Explaining the Predictions of Any Classifier paper. The authors of the paper have developed a Python implementation and while the two implementations share many overlaps, the R package is not a direct port of the Python code, rather it’s an implementation idiomatic to R, playing on the strength of the language. The most notable difference from a user point-of-view is the lack of support for explaining image predictions in the R version. This a transient situation though, as it will get added at a later stage.
The R lime implementation has been a joint effort between myself and Michaël Benesty, with Michaël handling the text prediction parts and me taking care of tabular data predictions.
Why would you explain a prediction?
Before going into the how of using lime, let’s talk a bit about the why. For many years models were often created just as much for understanding a system as for predicting new observations. Inspections and understanding of the models themselves were a way to understand and make sense of the system and thus a goal in itself. The recent surge in use of complex machine learning algorithms has shifted the expected need of a model towards the prediction side, as the models are often too complex to understand. Simply put, in order to explain more and more complex phenomena, the models must become increasingly complex. While the new crop of algorithms performs impressively in the prediction department, their black-box nature presents a real problem that has been swept under the carpet for long, as more and more accurate algorithms has been pursued. While you might convince yourself that you simply don’t care that much about the mechanics of the system, you’re simply interested in high quality predictions, this sentiment simply doesn’t cut it for several reasons:
- The end users of your prediction might have much less confidence in the infallibility of your algorithm and if you cannot explain how your model reaches a prediction they are likely to ignore your predictions. A prime example is a system predicting medical conditions requiring drastic surgery. In order for doctors to follow the predictions they must have knowledge of which factors weighted in.
- Laws and regulations are cropping up to restrict the blind use of complex models. Next year all EU countries must adhere to a new regulation requiring algorithmic decisions to be explainable to the citizens directly affected.
- Your setup might be flawed resulting in a model that performs well on your test and training data but in reality is a poor reflection of your problem. There are many ways this can happen and there are many stories of how it can lead to real implications for affected people, such as in the case of racist bias in algorithms performing risk assessments on defendants or models claiming to be able to predict sexual orientation from portraits.
- You might not have made the model you use and do not have access to how it was trained, thus you need to poke to it in order to understand how it behaves.
- Are you really sure you have no interest in understanding the system? You probably should begin to be interested in understanding the system! One of the best ways to improve models is to infuse them with domain knowledge and domain knowledge is what comes out of understanding the system.
How should you explain a prediction?
Having successfully convinced you that explaining model predictions is a valuable, even sometimes indispensable, tool, we must begin to discuss what constitutes an explanation. The authors of the lime article puts forth several points worth considering (and which have of course guided the development of the lime framework).
Locally stable
The complexity of current models are needed to overcome the non-linearity of the problems they are applied to solve. Thus, we cannot expect explanations to be globally stable, since what holds true for one observation might be false for another due to different mechanics a play n different areas of the feature space. Still, we must expect an explanation to be locally stable. If the slightest perturbation in an observation results in dramatic changes to how the prediction is explained, the explanation itself holds little value. Requiring explanations to be locally stable will allow us to gain insight into the neighborhood of explained observations.
Understandable
In order for an explanation to be useful it must be, well, understandable. Several factors weighs in when determining the understandability of an explanation. The level of expertise of the end recipient, the number of parameters used in the explanation, and the complexity of the relationship between the parameters. As an example, consider a classic linear model. This is generally considered to be a low-complexity model that is fairly understandable. Still, if it consists of 50 variables it might become incomprehensible due to the amount of interactions between variables etc.
Another part of understandability is how an explanation can be presented. This is especially important when the recipients of the explanation does not work with statistics as a dump of raw model parameters will mean nothing to them. Thus, an explanation must be able to be put in the context of the original input so that most people will get a sense of its implications.
Model-agnostic
While a single explanation in itself does not need to be model-agnostic it is a very important requirement if you seek to develop an approach to explaining models in general. If the explanation framework can treat the model as a black box it can be used on any type of model. This leads to three important advantages:
- Future proof If the framework doesn’t care about the inner workings of the model it is sure to continue to work with the models being developed in the future.
- Explanation consistency If the way predictions were explained varied from model to model, it would require a larger mental effort to switch between model prediction explanations.
- Confidentiallity proof If you do not own the model, and the model is in fact locked away from prying eyes, it is an absolute necessity to treat it as a black box.
Learn more
Fast Forwards Labs recently held a web seminar on machine learning interpretability which both touch upon lime directly as well as discusses the need for model interpretability. They have made a recording of this available which I would invite everyone to watch.
They have also compiled a list of additional resources at the seminar webpage which is a good starting point for learning more about the subject.
How do lime tackle these problems
Behind the workings of lime lies the (big) assumption that every complex model is linear on a local scale. It is not difficult to convince yourself that this is generally sound — you usually expect two very similar observations to behave predictably even in a complex model. lime then takes this assumption to its natural conclusion by asserting that it is possible to fit a simple model around a single observation that will mimic how the global model behaves at that locality. The simple model can then be used to explain the predictions of the more complex model locally.
The general approach lime takes to achieving this goal is as follows:
-
For each prediction to explain, permute the observation
ntimes. - Let the complex model predict the outcome of all permuted observations.
- Calculate the distance from all permutations to the original observation.
- Convert the distance to a similarity score.
-
Select
mfeatures best describing the complex model outcome from the permuted data. -
Fit a simple model to the permuted data, explaining the complex model outcome with the
mfeatures from the permuted data weighted by its similarity to the original observation. - Extract the feature weights from the simple model and use these as explanations for the complex models local behavior.
It is clear from the above that there’s much wiggle-room in order to optimize the explanation. Chief among the choices that influence the quality of the explanation is how permutations are created, how permutation similarity is calculated, how, and how many, features are selected, and which model is used as the simple model. Some of these choices are hard-coded into lime, while others can be influenced by the user — all of them will be discussed below.
How to permute an observation
When it comes to permuting an observation, lime depends on the type of input data. Currently two types of inputs are supported: tabular and text
-
Tabular Data When dealing with tabular data, the permutations are dependent on the training set. During the creation of the explainer the statistics for each variable are extracted and permutations are then sampled from the variable distributions. This means that permutations are in fact independent from the explained variable making the similarity computation even more important as this is the only thing establishing the locality of the analysis.
-
Text Data When the outcome of text predictions are to be explained the permutations are performed by randomly removing words from the original observation. Depending on whether the model uses word location or not, words occurring multiple times will be removed one-by-one or as a whole.
Calculating similarities with permutations
Just as permutations are created differently based on the input data, so the similarities are calculated in different ways. For text data the cosine similarity measure is used, which is the standard in text analysis (it effectively measures the angle difference between the two feature vectors). For tabular data a bit more thought is required and the optimal solution will depend on the type of input data. First, the categorical features will be recoded based on whether or not they are equal to the observation. Second, if continuous features are binned (the default) these features will be recoded based on whether they are in the same bin as the observation. Using the recoded data the distance to the original observation is then calculated based on a user-chosen distance measure (euclidean by default), and converted to a similarity using an exponential kernel of a user defined width (defaults to 0.75 times the square root of the number of features).
Selecting the features to use
Feature selection is a complete sub-field of modelling in itself and lime has no silver bullet for this. Instead, it implements a range of different feature selection approaches that the user is free to choose from. First though, the number of features needs to be chosen. The number must strike a balance between the complexity of the model and the simplicity of the explanation, but try to keep it below 10 (personal opinion). As for selecting the features, lime supports the following algorithms:
- none: Use all features for the explanation. Not advised unless you have very few features.
- forward selection: Features are added one by one based on their improvements to a ridge regression fit of the complex model outcome.
-
highest weights: The
mfeatures with highest absolute weight in a ridge regression fit of the complex model outcome are chosen. -
lasso: The
mfeatures that are least prone to shrinkage based on the regularization path of a lasso fit of the complex model outcome is chosen. -
tree: A tree is fitted with
log2(m)splits, to use at maxmfeatures. It may possibly select less. -
auto: Uses forward selection if
m <= 6and otherwise highest weights.
Fitting a model to the permuted and feature-reduced data
Once permutations have been created, similarities calculated and features selected it is time to fit a model. If the complex model is a regressor, the simple model will predict the output of the complex model directly. If the complex model is a classifier, the simple model will predict the probability of the chosen class (for classifiers it is possible to either specify the classes to explain, or let lime chose the top k most probable classes).
The only requirement for the simple model is that it can work with weighted input and that it is easy to extract understanding from the resulting fit. While multiple types of models support this, lime uses a ridge regression as the model of choice. In the future this might be expanded to other types of models.
An example
So, this has been more text heavy than my usual post, but I hope you have gotten through it. A proper understanding of the theory and mechanics of lime is key to using it effectively. Still, as this is a software release note and not an academic article I will show you how to use it to just help you get your feet wet.
In the following, we are going to make a classification model with the aim of predicting whether a tumor is benign or malignant based on a number of measurements. In actuality we are fitting a linear discriminant model to the biopsy data set from MASS.
library(lime)
library(MASS)
data(biopsy)
# First we'll clean up the data a bit
biopsy$ID <- NULL
biopsy <- na.omit(biopsy)
names(biopsy) <- c('clump thickness', 'uniformity of cell size',
'uniformity of cell shape', 'marginal adhesion',
'single epithelial cell size', 'bare nuclei',
'bland chromatin', 'normal nucleoli', 'mitoses',
'class')
# Now we'll fit a linear discriminant model on all but 4 cases
set.seed(4)
test_set <- sample(seq_len(nrow(biopsy)), 100)
prediction <- biopsy$class
biopsy$class <- NULL
model <- lda(biopsy[-test_set, ], prediction[-test_set])We now have our model, and we can see how well it performs
sum(predict(model, biopsy[test_set, ])$class == prediction[test_set])/100## [1] 0.96
So, it’s pretty accurate. But lets see how these predictions came to be, using lime. Usage of lime is split in two operations. First, you train an explainer for your model (using the lime() function), which is kept around alongside your model. Then new observations are predicted and explained using the explain() function.
# Train the explainer
explainer <- lime(biopsy[-test_set,], model, bin_continuous = TRUE, quantile_bins = FALSE)
# Use the explainer on new observations
explanation <- explain(biopsy[test_set[1:4], ], explainer, n_labels = 1, n_features = 4)
tibble::glimpse(explanation)## Observations: 16
## Variables: 13
## $ model_type <chr> "classification", "classification", "classifi...
## $ case <chr> "416", "416", "416", "416", "7", "7", "7", "7...
## $ label <chr> "benign", "benign", "benign", "benign", "beni...
## $ label_prob <dbl> 0.9964864, 0.9964864, 0.9964864, 0.9964864, 0...
## $ model_r2 <dbl> 0.5659044, 0.5659044, 0.5659044, 0.5659044, 0...
## $ model_intercept <dbl> 0.08837631, 0.08837631, 0.08837631, 0.0883763...
## $ model_prediction <dbl> 1.0244738, 1.0244738, 1.0244738, 1.0244738, 0...
## $ feature <chr> "normal nucleoli", "bare nuclei", "uniformity...
## $ feature_value <int> 5, 3, 3, 3, 1, 10, 1, 1, 5, 10, 10, 3, 1, 1, ...
## $ feature_weight <dbl> -0.018041571, 0.573050022, 0.202345467, 0.178...
## $ feature_desc <chr> "3.25 < normal nucleoli <= 5.50", "bare nucle...
## $ data <list> [[3, 3, 2, 6, 3, 3, 3, 5, 1], [3, 3, 2, 6, 3...
## $ prediction <list> [[0.9964864, 0.003513577], [0.9964864, 0.003...A problem with these explanations is that the local model does not provide a very good fit. We can try to improve it by changing the kernel width, effectively changing the size of the neighborhood it is fitting around.
explanation <- explain(biopsy[test_set[1:4], ], explainer, n_labels = 1,
n_features = 4, kernel_width = 0.5)
explanation[, 2:9]## case label label_prob model_r2 model_intercept model_prediction
## 1 416 benign 0.9964864 0.4804993 0.4323631 1.0029394
## 2 416 benign 0.9964864 0.4804993 0.4323631 1.0029394
## 3 416 benign 0.9964864 0.4804993 0.4323631 1.0029394
## 4 416 benign 0.9964864 0.4804993 0.4323631 1.0029394
## 5 7 benign 0.9244742 0.4680113 0.3216358 0.6370384
## 6 7 benign 0.9244742 0.4680113 0.3216358 0.6370384
## 7 7 benign 0.9244742 0.4680113 0.3216358 0.6370384
## 8 7 benign 0.9244742 0.4680113 0.3216358 0.6370384
## 9 207 malignant 0.9999911 0.6543314 0.1583423 1.0001967
## 10 207 malignant 0.9999911 0.6543314 0.1583423 1.0001967
## 11 207 malignant 0.9999911 0.6543314 0.1583423 1.0001967
## 12 207 malignant 0.9999911 0.6543314 0.1583423 1.0001967
## 13 195 benign 0.9999981 0.4631399 0.5493353 1.0047116
## 14 195 benign 0.9999981 0.4631399 0.5493353 1.0047116
## 15 195 benign 0.9999981 0.4631399 0.5493353 1.0047116
## 16 195 benign 0.9999981 0.4631399 0.5493353 1.0047116
## feature feature_value
## 1 mitoses 1
## 2 bare nuclei 3
## 3 clump thickness 3
## 4 uniformity of cell size 3
## 5 mitoses 1
## 6 bare nuclei 10
## 7 clump thickness 1
## 8 uniformity of cell size 1
## 9 mitoses 1
## 10 uniformity of cell size 10
## 11 clump thickness 10
## 12 uniformity of cell shape 9
## 13 mitoses 1
## 14 bare nuclei 1
## 15 clump thickness 3
## 16 uniformity of cell size 1This improved it somewhat, but it could probably become even better.
A more powerful way of looking at the explanations is with the graphical overview provided by the plot_features() function.
plot_features(explanation, ncol = 1)
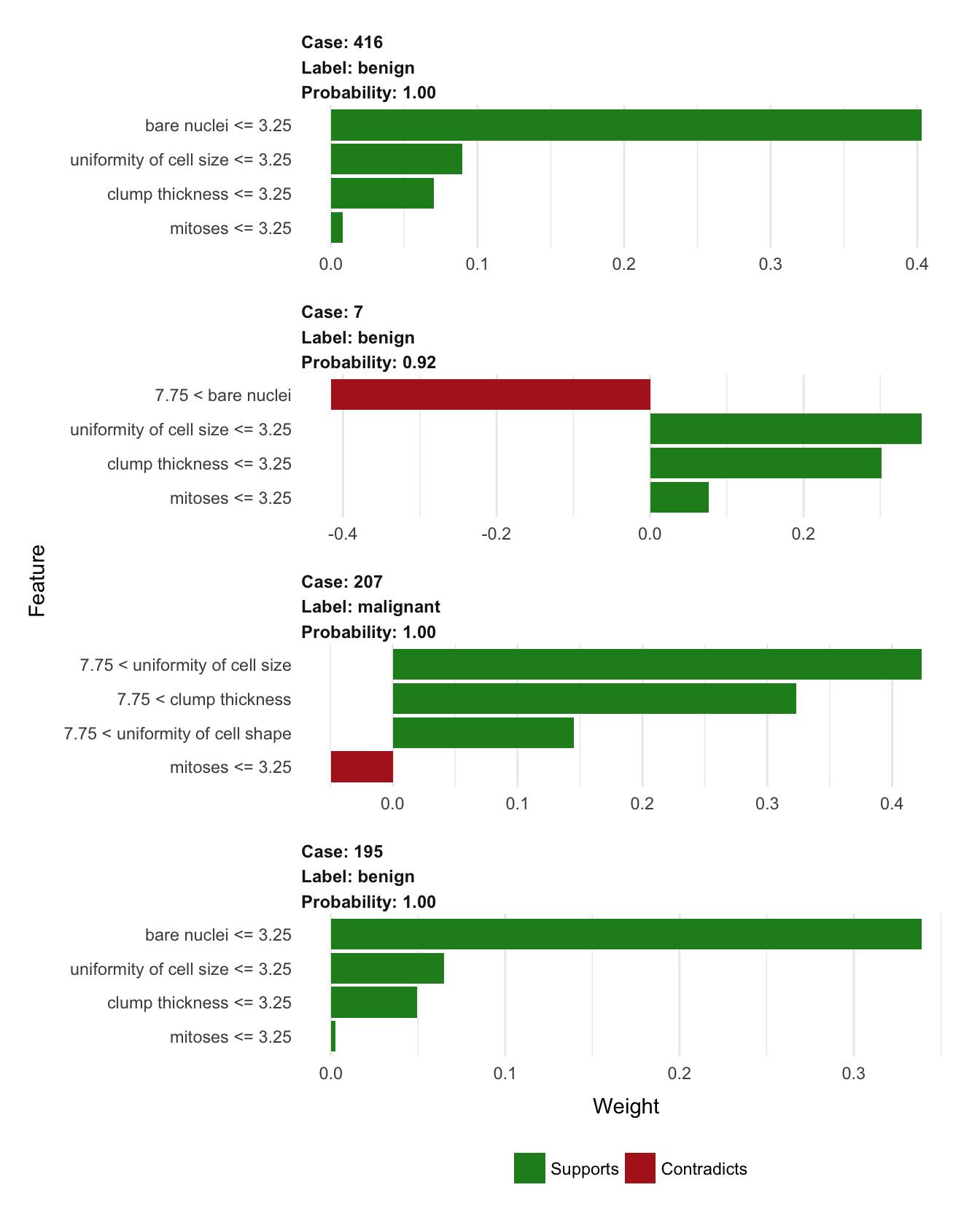
In this overview it is clear to see how case 195 and 416 behave alike, while the third benign case (7) has an unusual large bare nuclei which are detracting from its status as benign without affecting the final prediction (indicating that the values of its other features are making up for this odd one). To no surprise it is clear that high values in the measurements are indicative of a malignant tumor.
What the future holds…
With this release we mark a point of stability for the lime API. The next goal will be to get feature parity with the Python version, which effectively means to support image predictions. Apart from that it is the goal to get as much user experience and feedback as possible in order to improve upon the methodology. We have no goal of sticking with the Python version if new and exciting ideas present themselves. So, hopefully I have convinced you of taking lime for a spin and if you do, I would love some feedback.